Enfermedad de Parkinson
Introducción y epidemiología
Descrita en 1817 por James Parkinson, la enfermedad de Parkinson es una enfermedad neurodegenerativa, crónica y progresiva que produce principalmente alteraciones en el sistema motor. La enfermedad de Parkinson es la causa más frecuente de parkinsonismo (temblor de reposo, rigidez, bradicinesia e inestabilidad postural) (Tolosa 2006, Olanow 2001)
El mal de Parkinson se desarrolla con mayor frecuencia después de los 50 años de edad y es uno de los trastornos neurológicos más comunes en los adultos mayores. En ocasiones, se presenta en adultos más jóvenes y afecta tanto a hombres como a mujeres.
La prevalencia de esta enfermedad es de 0,3% en la población general y de 1% en mayores de 60 años. La edad promedio de inicio es 60 años con una sobrevida promedio desde el diagnóstico de unos 15 años. La causa de muerte es difícil de identificar en la mayoría de los casos; de las causas identificables, la más frecuente es neumonía. Su incidencia aumenta con la edad desde 17,4/100000 habitantes por año entre los 50-59 años a 93,1/100000 habitantes por año entre los 70-79 años. El riesgo de desarrollar la enfermedad es de un 1,5% a lo largo de la vida. Es 1,5 veces más frecuente en hombres que en mujeres. (Nussbaum 2003, de Lau 2006, Lees 2009)
En algunos casos, la enfermedad es hereditaria. Cuando una persona joven resulta afectada, generalmente se debe a una forma de la enfermedad que es hereditaria.
Las neuronas utilizan un químico cerebral, llamado dopamina, para ayudar a controlar el movimiento muscular. El mal de Parkinson ocurre cuando las neuronas del cerebro que producen la dopamina se destruyen lentamente. Sin la dopamina, las neuronas en esa parte del cerebro no pueden enviar mensajes apropiadamente, llevando a la pérdida de la función muscular. El daño empeora con el tiempo. Se desconoce la razón exacta por la cual las neuronas se desgastan. (Nussbaum 2003, de Lau 2006, Lees 2009).
El uso de levodopa (medicamento utilizado para esta enfermedad) da a estos pacientes una expectativa de vida casi normal. (Hoehn y Yahr, 1967, Marttila 1977)
Patogénesis
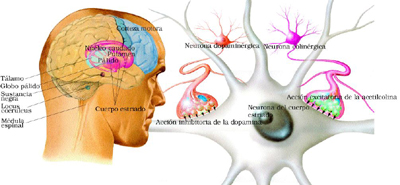
La modulación del movimiento a nivel de los ganglios basales, se produce a partir de la sustancia negra (parte compacta) que actúa mediante la dopamina a través de dos vías; una indirecta a través del cuerpo estriado- globo pálido (porción externa) – núcleo subtalámico – globo pálido (porción interna) – tálamo que ejerce una función inhibitoria finalmente en la corteza; y otra vía directa a través del cuerpo estriado – globo pálido (porción interna) – tálamo, que ejerce una función excitatoria a nivel cortical (Figura 1). (Gerfen 2000)
En la enfermedad de Parkinson se produce una depleción dopaminérgica, en la sustancia negra y en la via nigro-estriatal hacia el cuerpo estriado. Al disminuir la dopamina en la sustancia negra se produce una hipersensibilidad de los receptores de dopamina D2 y una hiporespuesta de los receptores D1 en el cuerpo estriado. Esto genera una sobre estimulación de la vía indirecta e inhibición de la vía directa lo que finalmente produce una inhibición de la corteza motora, lo que se manifiesta clínicamente en síntomas parkinsonianos típicos como la bradicinesia. (Bamford 2004) ( Figura 1)


Figura 1: A la Izquierda se observa el circuito ganglionar normal. En verde está señalada la vía indirecta, a partir del cuerpo estriado con sus receptores D2. En amarillo está señalada la vía directa a partir de los receptores D1. A la derecha está representada la enfermedad de Parkinson donde por disminución de la dopamina en la sustancia negra se produce una hiper reactividad de los receptores D2 e hipo reactividad de los receptores D1. Esto produce una sobre estimulación de la vía indirecta por sobre la vía directa; lo que finalmente inhibe el impulso excitatorio del tálamo hacia la corteza motora, generando la pobreza de movimiento.
El mecanismo neurodegenerativo preciso que genera esta enfermedad no está claro, sin embargo diferentes eventos (genéticos, medio ambientales, inflamatorios, inmunológicos, estrés oxidativo, etc.) interactuarían entre sí produciendo finalmente apoptosis y necrosis de las neuronas de la sustancia negra. (Cory-Slechta 2005, Schulz 2008)
Histopatológicamente se puede observar despigmentación (por disminución de
neuromelanina), pérdida neuronal y gliosis, principalmente en la sustancia negra parte compacta y locus ceruleus (puente). En las neuronas restantes se puede observar los Cuerpos de Lewy; que corresponden a inclusiones eosinofílicas intranucleares, características de la enfermedad de Parkinson. (Spillantini 1997)
El principal factor de riesgo para desarrollar esta enfermedad neurodegenerativa es la edad, habiendo sólo un 10% de casos menores de 45 años. También se han postulado otros factores de riesgo como exposición laboral a pesticidas, metales pesados, el sobrepeso y dietas altas en fierro; sin embargo la evidencia al respecto no es concluyente. También se ha visto una mayor frecuencia de enfermedad de Parkinson en algunas enfermedades metabólicas (Ej: Enf. Gaucher) siendo más frecuente esta asociación en la población judía Ashkenazi. (de Lau 2006)
Además se ha atribuido un rol protector para el tabaco, ya que se ha visto que aquellas personas que nunca han fumado tienen el doble de riesgo de desarrollar esta enfermedad. Por otro lado se ha encontrado una menor incidencia en personas que consumen cafeína. (Ritz 2007, Ross 2000)
Si bien la mayoría de los casos son esporádicos, hay formas genéticas de esta enfermedad. Especialmente cuando son de presentación temprana (menores de 50 años). Determinan edad de presentación más temprana, con un curso más benigno y raramente presentan síntomas bulbares o demencia. (Gasser T 2007, Lees 2009).
Síntomas:
El trastorno puede afectar uno o ambos lados del cuerpo. La magnitud de la pérdida funcional puede variar.
Los síntomas pueden ser leves al principio. Por ejemplo, el paciente puede tener un temblor leve o una ligera sensación de que una pierna o pie está rígido y se arrastra.
Los síntomas abarcan:
- Movimientos automáticos (como parpadear) que disminuyen o se detienen
- Estreñimiento
- Dificultad para deglutir
- Babeo
- Alteración del equilibrio y la marcha
- Falta de expresión facial (aspecto de “máscara”)
- Achaques y dolores musculares
- Problemas con el movimiento
- dificultad para iniciar o continuar un movimiento, como comenzar a caminar o pararse de una silla
- pérdida de movimientos pequeños o finos de la mano (la escritura puede volverse pequeña y difícil de leer; comer se vuelve más difícil)
- movimientos lentos
- postura encorvada
- Músculos rígidos o tensos (a menudo comenzando en las piernas)
- Agitación, temblores
- generalmente ocurren en las extremidades en momentos de reposo o cuando se extiende el brazo o la pierna
- los temblores desaparecen durante el movimiento
- con el tiempo, el temblor se puede observar en la cabeza, los labios, la lengua y los pies
- pueden empeorar cuando la persona está cansada, excitada o estresada
- puede presentarse frotamiento del pulgar y dedos de la mano (rodamiento de píldora)
- Habla más tranquila y lenta, y voz monótona
Otros síntomas:
- Ansiedad, estrés y tensión
- Confusión
- Demencia
- Depresión
- Desmayo
- Alucinaciones
- Pérdida de la memoria
Manifestaciones clínicas
Los síntomas cardinales son la bradicinesia, el temblor y la rigidez. También se considera dentro de estos la inestabilidad postural, pero ésta es de inicio más tardío. Su presentación es gradual y muchas veces sus síntomas atribuidos a la edad, lo que produce un retraso en el diagnóstico de 2 a 3 años. (Gelb 1999, Ward 1990, Hughes 1992)
El temblor es de reposo, disminuye al mantener una postura o realizar un movimiento. Presente en el 70% de los pacientes con Parkinson, generalmente se inicia en una mano con compromiso unilateral para luego extenderse al lado contralateral a lo largo de los años. Puede afectar también piernas, lengua, labios y mandíbula. (Hoehn 1967, Scout 1970, Hunker 1990, Chou 2009)
La bradicinesia se define como una lentitud en el inicio, realización o finalización del movimiento voluntario. Es la principal causa de discapacidad en estos pacientes. Afecta prácticamente a todos los pacientes. En el brazo parte de distal a proximal, con deterioro de la motricidad fina inicialmente, también se observa disminución o ausencia de braceo. En las piernas se manifiesta por alteración en la marcha con enlentecimiento y pasos cortos además de sensación de inestabilidad. Cuando la enfermedad progresa, el congelamiento de la marcha y la festinación se presentan. James Parkinson definió la festinación como “un irresistible impulso ha hacer los pasos muchos más cortos y rápidos” (Parkinson 1817)
La rigidez, que está presente en el 90% de los pacientes, generalmente es de inicio unilateral y en el mismo lado del temblor. Puede progresar al lado contralateral pero se mantiene de forma asimétrica. El lado de inicio sigue siendo el más afectado. Puede presentarse en forma de rueda dentada. Esto significa que hay breves episodios de oposición alternados con episodios de relajación. En otros casos la resistencia se mantiene constante, lo que se denomina “rigidez en tubo de plomo”. Aquí la resistencia persiste uniforme en todo el rango del movimiento, tanto en flexión como en extensión (tubo de plomo) y no cambia al variar la velocidad con que se moviliza el segmento, a diferencia de la espasticidad. (Hughes 1992, Scott 1970, Deuschl 1998, Chou 2009, Ministerio de salud 2007)
Las alteraciones posturales son más tardías y se manifiestan por inestabilidad con tendencia a las caídas. (Muslimovic 2008). Puede haber, además, facie hipomímica, hipofonía, disfagia, alteración del parpadeo, micrografía y distonías.
Si bien esta enfermedad afecta principalmente al sistema motor, también puede tener manifestaciones no motoras y neuro-psiquiátricas como alteraciones cognitivas, psicosis, trastornos del ánimo, del sueño, disfunciones autonómicas (ortostatismo, constipación, incontinencia urinaria) y olfatorias (hiposmia). (Langston 2006, Chou 2009)
Diagnóstico
El diagnóstico es clínico, debe presentar al menos 2 de las manifestaciones cardinales sumado a una respuesta satisfactoria a la terapia con levodopa. El diagnóstico es más probable si además tiene inicio unilateral, temblor de reposo o asimetría durante la evolución siendo el lado inicial el más comprometido. (Hughes 1992)
Debe tenerse presente, como diagnóstico diferencial, otras enfermedades neurodegenerativas que también se presentan con síntomas similares, como la demencia por cuerpos de Lewy, la degeneración corticobasal, la atrofia multisistémica y la parálisis supranuclear progresiva. (Tolosa 2006, Ahlskog 2000)
Debe descartarse otras condiciones que producen parkinsonismo secundario, dentro de las cuales está el uso de drogas (como antipsicóticos, antieméticos, bloqueadores del calcio), la exposición a tóxicos (como el monóxido de carbono y metanol) y las lesiones estructurales (tumores, hidrocefalia, hematoma subdural crónico, infecciones del SNC, enfermedad cerebrovascular) (Chou 2009, Tolosa 2006, Mena 2006, Ministerio de salud 2007)
En general debe buscarse otra causa de parkinsonismo si las caídas se presentan al inicio de la enfermedad, si tiene una mala respuesta a levodopa, si hay simetría al inicio, progresión rápida, disautonomía al inicio de la enfermedad o ausencia de temblor. En caso de sospechar causas secundarias podría requerirse una neuroimagen. (Chou 2009, Suchowersky 2006, Ministerio de salud 2007)
Tratamiento en la Enfermedad de Parkinson
El tratamiento debería ser integral, a través de un equipo multidisciplinario, mantenerse a lo largo de la vida y contar con apoyo familiar y social. El manejo idealmente debe abordar medidas farmacológicas y no farmacológicas.(Carter 1998, Cornella 1994)
Esta revisión sólo abordará el manejo farmacológico de los síntomas motores de la Enfermedad de Parkinson. El tratamiento actualmente disponible es sintomático. La decisión de ocupar un fármaco sobre otro, así como el momento de iniciar la terapia, debe ser individualizada. Esto tomando en cuenta la edad, el impacto de la enfermedad, los posibles efectos adversos y los costos relacionados con la medicación. (Olanow 2001)
La droga cardinal en el tratamiento de la enfermedad de Parkinson es la levodopa; una prodroga precursora de la dopamina. Es el fármaco más efectivo. Se ha asociado a una menor morbimortalidad en comparación con la era pre-levodopa y prácticamente beneficia a todos los pacientes con enfermedad de Parkinson confirmada. Se usa en combinación con un inhibidor de la descarboxilasa (carbidopa o benserazida), para prevenir los síntomas propios de su conversión periférica a dopamina (náuseas, vómitos e hipotensión ortostática). (Olanow 2001, Diamond 1989).
A pesar de la efectividad de la levodopa, su uso se ha limitado por un posible rol neurotóxico. En estudios in vitro se ha visto que el uso de levodopa aumentaría la liberación de radicales libres en las neuronas dopaminérgicas produciendo muerte celular. Sin embargo, la concentración de levodopa usada en estos estudios es considerablemente mayor que la alcanzada en los pacientes con Parkinson. Además los cultivos de neuronas dopaminérgica carecen de los mecanismos de defensa proporcionados por las células gliales in vivo. (Stocchi 2005, Basma 1995, Benetello 1993, Mena 1992)
En el estudio ELLDOPA, se demostró que la levodopa tenía un menor deterioro de la condición basal en la UPDRS (Unified Parkinson’ s Disease Ranking Scale) en comparación a placebo; lo que no apoyaría un rol tóxico de la levodopa. Sin embargo en el mismo estudio, el análisis de neuroimágenes, demostró que en los pacientes tratados con levodopa, había una mayor tasa de declinación en biomarcadores de la función nigroestratia. (Fahn at al 2004)
Incluso en el consenso realizado sobre este tema, se llegó a la conclusión de que no existe suficiente evidencia actual que apoye un efecto tóxico de la levodopa en las neuronas nirgroestratiales, por lo que este tema persiste en controversia. (Agid 1998).
El principal inconveniente del uso prolongado de levodopa es la aparición de complicaciones motoras. Dentro de éstas está el fenómeno de ‘wearing off’; que corresponde a una reaparición o empeoramiento de los síntomas parkinsonianos, antes de que haga efecto la siguiente dosis de levodopa. Se puede acompañar de
La aparición de las complicaciones motoras ocurre aproximadamente en el 50-90% de los pacientes que han recibido levodopa por 5 a 10 años, porcentaje que aumenta hasta el 100% en pacientes con enfermedad de Parkinson de inicio temprano. Estas complicaciones se relacionan con una mayor concentración plasmática de levodopa. (Hauser 2006, Schrag 2000)
La decisión de cuando iniciar el tratamiento sintomático es controversial. Se ha planteado su uso al inicio de la enfermedad para alcanzar un mayor beneficio clínico, sin embargo otros prefieren atrasar su uso debido a las complicaciones motoras o por el potencial rol neurotóxico. En el estudio DATATOP el punto de inicio del tratamiento fue cuando los pacientes referían dificultad en la realización
Finalmente la decisión del inicio de la terapia debiera ser individualizada, con la menor dosis posible, principalmente determinada por el deterioro funcional del paciente. (Parkinson Study Group 1996, Olanow 2001).
Debido a estas limitaciones han aparecido otras opciones farmacológicas. Éstas son los agonistas dopaminérgicos (AD), los inhibidores de la monoamino oxidasa tipo B (MAO B), los inhibidores de la catecol-o-metil transferasa (COMT), los anticolinérgicos (AC) y la Amantadina.
Los agonistas dopaminérgicos, dentro de los cuales están la bromocriptina, el pramipexol, el ropinirol, la rotigotina y la apomorfina inyectable, estimulan directamenteel receptor de dopamina. Su uso como monoterapia en comparación con levodopa ha mostrado una menor incidencia de complicaciones motoras, pero un menor control sintomático y mayores efectos adversos. En una revisión sistemática publicada en el 2008, los pacientes tratados con AD tuvieron un 50% menos de discinesia, un 40% menos de distonía y un 25% menos de fluctuaciones motoras que aquellos tratados con levodopa. Los principales efectos adversos publicados en este estudio como edema, somnolencia, constipación, mareos, alucinaciones y naúseas, fueron más frecuentes en los pacientes tratados con AD. En esta revisión, solo 4 estudios tenían buena calidad metodológica para evaluar el control sintomático, los que concluyeron que era mejor en el grupo de levodopa. (Stowe 2008).
En el estudio CALM-PD que midió el efecto a largo plazo del inicio de pramipexol versus levodopa en etapa temprana de la enfermedad de Parkinson, dio como resultado una frecuencia de 68,4% de complicaciones motoras en el grupo de levodopa versus un 50% en el grupo de pramipexol (p<0.02), mientras que la somnolencia fue mayor en el grupo de pramipexol. No hubo diferencias significativas en el control de síntomas y la calidad de vida (Parkinson Study Group 2009).
El uso combinado de agonistas dopaminérgicos más levodopa en comparación al uso de levodopa sola, tendría mejor control sintomático, con menores complicaciones motoras y una reducción de de la dosis de levodopa. (Talati 2009)
Son escasos los estudios que han comparado la eficacia de los agonistas dopaminérgicos entre sí; y no han encontrado diferencia significativa entre ellos. (Guttman 1997, Pezzoli 1994).
Los AD ergotamínicos (cabergolina y pergolida) se han asociados con riesgo de enfermedad valvular cardiaca, por lo que se aconseja el uso de los no ergotamínicos , ya que tienen similar perfil de eficacia. (Zanettini 2007).
Los inhibidores de la MAO B actúan disminuyendo la inactivación de la dopamina a nivel del sistema nervioso central. Sólo está disponible en Chile la selegilina. Su efecto sintomático es modesto, y su uso como monoterapia tiene escaso control sintomático, sin embargo asociación a levodopa ha mostrado ser mejor que el uso de levodopa sola. (Horn at al 2004, Palhagen 2006)
En el 2004 se publicó una revisión que comparó iMAO B, principalmente selegilina, con o sin levodopa, versus placebo y levodopa. Este estudio fue realizado en pacientes en etapa temprana de la enfermedad, definida como aquella que no ha tenido complicaciones motoras, que no ha sido tratada o que haya recibido menos de un año de tratamiento farmacológico. Esta revisión mostró que el uso de selegilina retrasaría la necesidad de levodopa y cuando se dan asociados, se requieren menores dosis de ésta (67 mg menos). Elcontrol sintomático de la selegilina fue superior al placebo en los 3 primeros meses, pero su utilidad a largo plazo es discutible. Su efecto en el control de las fluctuaciones motoras es modesto. (Ives 2004).
Los inhibidores de la COMT disminuyen la metilación de la levodopa y la dopamina, aumentando su vida media plasmática. Estos son la entacapona y la tolcapona. Son inefectivos como monoterapia. Sin embargo su uso asociado a levodopa es beneficioso en el control de los síntomas motores, principalmente en pacientes que experimentan fenómenos de ‘wearing off’, mejorando la calidad de vida. (Nutt 1998, Olanow 2004).
En los ganglios basales la dopamina y la acetilcolina se encuentran en equilibrio. En la enfermedad de Parkinson, la disminución de dopamina genera un estado dehipersensibilidad colinérgica. Esto determina que las drogas con acción colinérgica
exacerben los síntomas de la enfermedad, mientras que las drogas anticolinérgicas los mejoran. Los anticolinérgicos disponibles en Chile son el trihexifenidilo y el biperideno. Como monoterapia o en asociación a otras drogas antiparkinsonianas tienen mejor control de síntomas motores que el placebo, sin embargo tienen una mayor frecuencia de alteraciones cognitivas y neuropsiquiátricas; lo que frecuentemente determina el abandono del tratamiento. (Katzenschlager 2003).
La amantadina es un antiviral con propiedades antiparkinsonianas, cuyo mecanismo de acción no está claro. Estudios en la década del 70 mostraron beneficios en el control de síntomas motores como monoterapia o en asociación con levodopa. (Parkes 1974, Eschwab 1972).
En referencia al contexto nacional, el piloto de la guía clínica GES para Enfermedad de Parkinson (2007) garantiza acceso a los siguientes medicamentos que se entregarán a nivel de atención primaria, ellos son: levodopa-carbidopa, levodopa-benzerasida, pramipexol, trihexifenidilo y quetiapina. (Ministerio de Salud 2007)
En la tabla 1 se presentan las principales drogas utilizadas en el tratamiento de la
enfermedad de Parkinson, los efectos adversos más frecuentes y algunas consideraciones que hay tener presente.
Tabla 1: Drogas utilizados en enfermedad de Parkinson y sus principales efectos adversos.
| Droga | Dosis diaria | Efectos adversos | Observaciones |
| Levodopa | 150-1500 mg | Nauseas, somnolencia, cefalea, mareos, alucinaciones, confusión, agitación, psicosis | No se recomienda dar con fenotizinas y metoclopramida |
| Selegilina | 0.5 mg | Nauseas, cefalea, insomnio, confusión. | No se recomienda dar con amitriptilina y antidepresivos IRSS |
| Pramipexol | 0.725-4.5 mg | Nauseas, vómitos, hipotensión ortostática, somnolencia, confusión, alucinaciones, edema periférico, constipación, mareos. | |
| Tolcapona | 300 mg | Discinecias, alucinaciones, confusión, nauseas, hipotensión ortostática, diarrea, alteración de p. hepáticas (tolcapona) | |
| Entacapona | 200 mg | Discinecias, alucinaciones, confusión, nauseas, hipotensión ortostática, diarrea, alteración de p. hepáticas (tolcapona) | |
| Trihexifenidilo | 1-6 mg | Alt memoria, confusión, alucinaciones, boca seca, visión borrosa, constipación, nauseas, retención urinaria, taquicardia. | Precaución si presenta Hiperplasia prostática benigna o glaucoma de ángulo estrecho y mistenia gravis. |
| Amantadina | 200-400 mg | Livedo reticularis, edema de extremidades inferiores, confusión, alucinaciones. | Precaución si presenta insuficiencia renal crónica. |

Conclusiones:
El tratamiento de la Enfermedad de Parkinson es individual y debe equilibrar el control de los síntomas motores versus los efectos secundarios de los fármacos.
La evidencia actualmente disponible apoya iniciar levodopa con o sin otro fármaco asociado, en pacientes con un importante deterioro funcional. En el resto de los pacientes y dependiendo de la edad podría usarse un agonista dopaminérgico de modo de atrasar las complicaciones motoras; que se asocian principalmente al uso prolongado y a altas dosis de levodopa.
Los iMAO B pueden utilizarse como monoterapia en etapas precoces y leves de la enfermedad o para control de fluctuaciones motoras secundarias al uso de levodopa.
Los iCOMT sólo deben indicarse asociados a levodopa y en etapas precoces de la enfermedad para disminuir las dosis de levodopa.
Finalmente el uso de anticolinérgicos y de la amantadina no poseen la evidencia suficiente como para proponerlos como drogas de primera línea.
Referencias Bibliográficas:
Agid Y, Chase T, Narsden D. Adverse reactions to levodopa: drug toxicity or progression of disease?. Lancet 1998 Mar 21; 351 (9106): 851-2.
Ahlskog JE. Diagnosis and differential diagnosis of Parkinson’s disease and parkinsonism. Parkinsonism Relat Disord. 2000 Nov 1;7(1):63-70.
Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004 Oct 27;24(43):9541-52.
Basma AN, Morris EJ, Nicklas WJ, Geller HM. L-DOPA cytotoxicity to PC12 cells in culture is via its autooxidation. J Neurochem 1995 Feb; 64 (2): 825-32.
Benetello P, Furlanut M, Zara G, Baroldo M, Hassan E. Plasma levels of levodopa and its main metabolites in parkinsonian patients after conventional and controlled-release levodopa-carbidopa associations. Eur Neurol 1993; 33:6973.
Carter JH,Stewart BJ, Archbold PG, Inoue I, Jaglin J, Lannon M, Rost-Ruffner E, Tennis M, McDermott MP, Amyot D, Barter R, Cornelius L, Demong C, Dobson J, Duff J, Erickson J, Gardiner N, Gauger L, Gray P, Kanigan B, Kiryluk B, Lewis P, Mistura K, Malapira T, Zoog K, et al. Living with a person who has Parkinson’s disease: the spouse’s perspective by stage of disease. Parkinson’s Study Group. Mov Disord 1998 Jan;13(1):20-8
Comella CL, Stebbins GT, Brown-Toms N, Goetz CG. Physical therapy and Parkinson’s disease: a controlled clinical trial. Neurology 1994 Mar;44(3 Pt 1):376-8.
Cory-Slechta DA, Thiruchelvam M, Barlow BK, Richfield EK. Developmental pesticide models of the Parkinson disease phenotype. Environ Health Perspect. 2005 Sep;113(9):1263-70
De Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006 Jun;5(6):525-35
Deuschl G, Bain P, Brin M .Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord. 1998;13 Suppl 3:2-23.
Diamond SG, Markham CH, Hoehn MM, Mc Dowel FH, Muenter MD. Effect of age at onset on progression and mortality in Parkinson’s disease. Neurology 1989 Sep; 39(9):1189-90
Fahns and the Parkinson Study Group. Levodopa and the progression of Parkinson disease. N Engl J Med 2004; 351:2498-2508.
Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord. 2007 Sep;22 Suppl 17:S343-50
Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999 Jan;56(1):33-9.
Gerfen CR. Molecular effects of dopamine on striatal-projection pathways. Trends
Neurosci. 2000 Oct;23(10 Suppl):S64-70.
Guttman, M. Double-blind comp arison of pramipexole and bromocriptine treatment with placebo in advanced Parkinson’s disease. Neurology 1997; 49:1060.
Hauser RA, Mc Dermontt MD, Messing S. Factors associated with the development of motor fluctuations and dyskinesias in Parkinson disease. Arch Neurol 2006 Dec, 631 (12): 1756-60.
Hillen ME, Sage JL. Nonmotor fluctuations in patients with parkinson’s disease. Neurology 1996; 47: 1180-1183.
Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967; 17:427.}
Hoehn, M. Yahr, M. Parkinsonism: onset, progression, and mortality. Neurology.1967; 17(5):427-442.
Horn, S, Stern, MB. The comparative effects of medical therapies for Parkinson’s disease. Neurology 2004; 63:S7
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992 Mar;55(3):181-4.
Hunker CJ, Abbs JH. Uniform frequency of parkinsonian resting tremor in the lips, jaw, tongue, and index finger. Mov Disord. 1990;5(1):71-7
Ives NJ, Stowe RL, Marro J, Counsell C, Macleod A, Clarke CE, Gray R, Wheatley K Monoamine oxidase type B inhibitors in early Parkinson’s disease: meta-analysis of 17 randomised trials involving 3525 patients BMJ Sep 2004; 329: 593; doi:10.1136/bmj.38184.606169.AE
Katzenschlager, R, Sampaio, C, Costa, J, Lees, A. Anticholinergics for symptomatic management of Parkinson’s disease. Cochrane Database Syst Rev 2003;:CD003735.
Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006 Apr;59(4):591-6.
Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet 2009 Jun 13;373(9680):2055-66.
Marttila, R. Rinne, U. Siirtola, T. Sonninen, V. Mortality of patients with parkinson’s disease treated with levodopa. J Neurol 1977; 216(3):147-153
Mena MA, de Yebenes JG. Drug-induced parkinsonism. Expert Opin Drug Saf. 2006 Nov;5(6):759-71.
Mena MA, Pardo B, Casarejos MJ, Fahns, García de Yebenest J. Neurototoxicity of levodopa on catecholemine-rich neurons. Mov Disord 1992; 7(1): 23-31. Ministerio de salud. Guía Clínica Enfermedad de Parkinson. Santiago: Minsal, 2007
Muslimovic D, Post B, Speelman JD, Schmand B, de Haan RJ. Determinants of disability and quality of life in mild to moderate Parkinson disease. Neurology. 2008 Jun 3;70(23):2241-7.
Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003 Apr 3;348(14):1356-64
Nutt, JG. Catechol-O-methyltransferase inhibitors for treatment of Parkinson’s disease. Lancet 1998; 351:1221.
Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009 May 26;72(21 Suppl 4):S1-136.
Olanow, CW, Kieburtz, K, Stern, M, et al. Double-blind, placebo-controlled study of entacapone in levodopa-treated patients with stable Parkinson disease. Arch Neurol 2004; 61:1563.
Olanow, CW, Watts, RL, Koller, WC. An algorithm (decision tree) for the management of Parkinson’s disease (2001): treatment guidelines. Neurology 2001; 56:S1.
Palhagen, S, Heinonen, E, Hagglund, J, et al. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006; 66:1200.
Parkes, JD, Baxter, RC, Marsden, CD, Rees, JE. Comparative trial of benzhexol,
amantadine, and levodopa in the treatment of Parkinson’s disease. J Neurol Neurosurg Psychiatry 1974; 37:422.
Parkinson Study Group CALM Cohort Investigators Long-term Effect of Initiating Pramipexole vs Levodopa in Early Parkinson Disease Arch Neurol. 2009 May;66(5):563-70.
Parkinson Study Group Levodopa and the progression of Parkinson’s disease. N Engl J Med 2004, Dec 9; 351 (24): 2498-2508.
Parkinson Study Group. Pramipexole vs Levodopa as Initial Treatment for Parkinson Disease: a 4-year randomized controlled trial. Arch Neurol. 2004 Jul;61(7):1044-53.
Parkinson Study Group. The impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requering levodopa. Ann Neurol 1996, 39: 23-36.
Parkinson, J. An Essay on the Shaking Palsy. Sherwood, Neely, and Jones, London 1817.
Pezzoli, G, Martignoni, E, Pacchetti, C, et al. Pergolide compared with bromocriptine inParkinson’s disease: A multicenter, crossover, controlled study. Mov Disord 1994; 9:431.
Strickland D, Van Den Eeden SK, Gorell J. Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol. 2007 Jul;64(7):990-7.
Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White LR Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000 May 24-31;283(20):2
674- 9.
Schrag A, Quinn N. Dyskinesias and motor fluctuations in Parkinoson’s disease. Brain 2000 Nov; 123 (Pt11): 2297-2305.
Schulz, J. Update on phatogenesis of Parkinson’s disease. J Neurol. 2008; 255 (suppl 5): 3-7
Schwab, RS, Poskanzer, DC, England, AC Jr, Young, RR. Amantadine in Parkinson’s disease. Review of more than two years’experience. JAMA 1972; 222:792.
Scott, RM, Brody, JA, Schwab, RS, Cooper, IS. Progression of unilateral tremor and rigidity in Parkinson’s disease. Neurology 1970; 20:710.
Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature 1997; 388:839.
Stocchi, F. Optimising levodopa therapy for the management of Parkinson’s disease J Neurol (2005) 252 [Suppl 4]: IV/43–IV/48 DOI 10.1007/s00415-005-4009-4 Stowe R, Ives N, Clarke CE, van Hilten, Ferreira J, Hawker RJ, Shah L, Wheatley K, Gray R. Dopamine agonist therapy in early Parkinson’s disease. Cochrane Database of Systematic Reviews 2008, Issue 2. Art. No.: CD006564. DOI: 10.1002/14651858.CD006564.pub2.
Suchowersky O, Reich S, Perlmutter J, Zesiewicz T, Gronseth G, Weiner WJ. Practice Parameter: diagnosis and prognosis of new onset Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006 Apr 11;66(7):968-75
Talati R, Baker W, Patel A, Reinhart K, Coleman C. Adding a dopamine agonist to preexisting levodopa therapy vs. Levodopa therapy alone in advanced Parkinson´s disease: a meta analysis. Int J Cli Pract, April 2009, 63, 4, 613-623.
Tarsy, D. Pharmacologic treatment of Parkinson disease. In: UpToDate, Basow, DS (Ed), UpToDate, Waltham, MA, 2009.
Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006 Jan;5(1):75-86
VanHilten, Ramaker CC, Stowe R, Ives N. Bromocriptine versus levodopa in early Parkinson’s disease. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No.:CD002258. DOI: 10.1002/14651858.CD002258.pub2.
Ward CD, Gibb WR. Research diagnostic criteria for Parkinson’s disease. Adv Neurol. 1990;53:245-9.
Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopa